
小小年纪大肚子,还伴有骨痛、贫血,谨防这种罕见病!
2025-02-19 梅斯罕见新前沿 MedSci原创 发表于陕西省

13 岁男孩小宇因肝脾肿大、骨痛等确诊戈谢病。该病是常见溶酶体贮积病,分多种类型,可通过酶活性测定等诊断,治疗有酶替代等方案,酶替代治疗对 1 型特异有效。
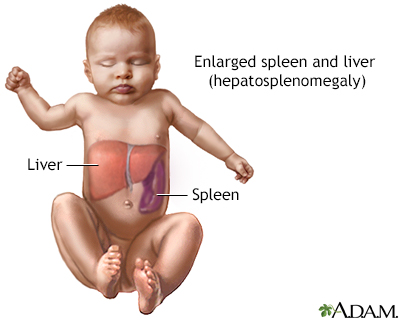
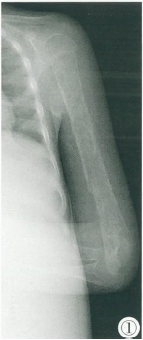
13岁的男孩小宇(化名)自出生以来,常常出现不同程度的肝脾肿大和骨痛,尤其是活动后,常抱怨背部和腿部疼痛。家人起初未重视,认为是生长发育过程中的正常现象。然而,随着年龄的增长,小宇的症状逐渐加重,特别是骨痛变得更加频繁且剧烈,甚至影响到正常的走路和跑步。最近几个月,小宇的脾脏和肝脏肿大明显,腹部常感到不适,且出现了轻度贫血和易疲劳的症状。家人开始感到担忧,带他多次就诊,最终在血液科医生的建议下,进行了酶活性检测和基因检查,确诊为戈谢病(GD)。
溶酶体贮积病
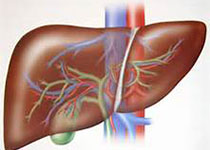
戈谢病(GD)是世界范围内最为常见的溶酶体贮积疾病之一,由于溶酶体中葡萄糖脑苷脂酶基因(GBA)突变导致其底物葡糖神经酰胺沉积在单核-巨噬细胞系统内,如肝、脾、肾等器官,甚至在脑的巨噬细胞溶酶体中贮积,并形成以“戈谢细胞”为特征的常染色体隐性遗传代谢病。
分型
根据神经系统是否受累,戈谢病主要分为非神经病变型(I型)和神经病变型(II型及III型)。还有其他少见的亚型如围生期致死型和心血管型等。
非神经病变型(即I型)患者最常见,约占95%,临床无原发性神经系统受累的症状,早期常出现轻度肝脾大伴脾功能亢进,严重可出现脾梗死、脾破裂等;可累及血液系统、呼吸系统、骨骼等。
急性神经病变型(即Ⅱ型)约1%,患病年龄较小,多数在出生后1年内发病,早期可死亡,神经系统的恶化呈急性发展。
亚急性神经病变型占2%~3%,发病多在青春期,神经系统受累进程相对缓慢,在未出现神经系统受累的症状时,早期与I型表现相似,很难与I型鉴别,寿命较长。
Ⅱ型和Ⅲ型是神经病变型,可出现神经系统受累的症状]。同时有大量的文献报道证实,关于GD患者恶性肿瘤的发病率呈现显著升高的趋势,甚至出现多种恶性肿瘤同时发生。
诊断
GD的诊断可通过酶活性测定(EAA),是通过测定白细胞或皮肤成纤维细胞中的葡萄糖脑苷酶活性其降低至正常值的30%以下,即可诊断为GD,是诊断GD的金标准。
戈谢细胞是GD的特征性表现,当镜下见到充满脂质的戈谢细胞,可高度怀疑GD,戈谢细胞的体积较大,形态不规则,多呈圆形、卵圆形,含有1个或数个偏位的细胞核,核染色质粗糙、致密,胞质量多,呈淡蓝色,胞质内充满交织成网状或洋葱皮样的条纹结构。
治疗
戈谢病的治疗有多种不同方案,包括酶替代治疗、脾脏切除以及支持疗法,其中酶替代治疗是目前戈谢病1型唯一特异有效的方法。
参考文献:
1.王艳霞,卫美辰,杨守京. 戈谢病3例临床病理学研究. 中华病理学杂志,2022,51(11):1158-1160.
2.霍双杰,连俊波,杨仕坤,等.成人Ⅰ型戈谢病1例报告及文献复习[J].诊断病理学杂志,2023,30(01):101-102.
3.李笛,陶晓娟,张宁宁,等.戈谢病患者肝脾病变定量磁共振影像研究[J].罕见病研究,2022,1(03):283-288.
 分享
分享
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#戈谢病# #溶酶体贮积病#
22 举报