
MOG抗体相关疾病(MOGAD) | 疑难探究
2025-06-20 神经科学论坛 神经科学论坛 发表于上海

MOGAD 是中枢神经系统自身免疫性疾病,以 MOG-IgG 为特征,儿童发病率更高,临床表现因年龄而异,可表现为视神经炎等,确诊依赖血清抗 MOG 抗体检测,治疗以高剂量 IV 皮质类固醇为主。
论坛导读:髓鞘少突胶质细胞糖蛋白抗体相关疾病(MOGAD)是是一种新型的中枢神经系统自身免疫性炎症性疾病,是一种既影响成人也影响儿童的罕见疾病,后者发病率更高,其临床表现因年龄而异。MOGAD与诸如多发性硬化(MS)和视神经脊髓炎谱系障碍(NMOSD)等其他神经炎性疾病有一些症状重叠,但这些疾病在病理生理学、预后和治疗反应方面存在重要差异。因此,需要对MOGAD进行准确和及时的诊断,以确保患者获得最佳治疗,并将因潜在可预防的疾病症状而住院的风险降至最低。
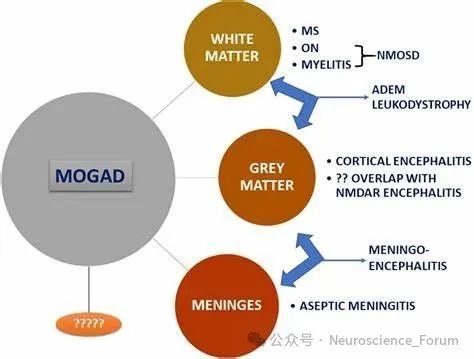
髓鞘少突胶质细胞糖蛋白抗体相关疾病(MOG antibody-associated disease,MOGAD)是一种中枢神经系统(CNS)疾病,其特征是存在以MOG为靶标的IgG自身抗体(MOG-IgG),以及影响儿童和成人视神经、脊髓、大脑或脑干的脱髓鞘病变。MOGAD是中枢神经系统自身免疫性炎性疾病中的一种新疾病,不同于多发性硬化(MS)和视神经脊髓炎谱系障碍(NMOSD)。尽管这三种疾病具有某些共同的临床特征,但它们在病理生理学、病程和治疗反应方面有所不同。MOGAD是一种既影响成人又影响儿童的罕见疾病,后者发生率更高。MOGAD的临床表现因年龄而异:在10岁以下的儿童中,经常出现急性播散性脑脊髓炎(ADEM ),而在10岁以上的儿童和成人中,更常观察到单侧或双侧视神经炎或急性脊髓炎。其他更罕见的表现也有报道,包括癫痫发作的脑炎表现。放射学发现有时有助于指导诊断:例如,广泛的前视神经受累、神经炎、累及脊髓圆锥的广泛脊髓病变和累及脑桥。通过测量血清中的抗MOG抗体来确诊。
MOGAD的平均发病年龄为28-30岁;然而,大约30%的MOGAD患者是儿童。因此,残疾和失明可能会持续几十年。虽然有些患者的病程是单相的,但约 40% 的成人和 30% 的儿童在发病后 5 年内复发,早期证据表明,更长时间的随访可能会发现更高的复发率。MOG IgG 血清阳性是诊断 MOGAD的先决条件。虽然相当一部分患者的病情会复发,但目前还没有能预测病程的生物标志物。非P42 MOG-IgG可预测相当一部分MOGAD患者的复发过程。单侧视神经炎是最常见的 MOGAD 表型,无论患者的年龄和性别如何,均可在发病时进行可靠的检测。对这些患者进行早期检测和专门治疗可以最大限度地减少残疾并改善长期预后。
MOGAD通常与急性播散性脑脊髓炎、视神经炎或横贯性脊髓炎相关,较少与大脑皮质脑炎、脑干表现或小脑表现相关。MOGAD可以表现为单相或复发性疾病过程,基于MOG-IgG细胞的检测对诊断准确性很重要。需要排除MS等诊断,但并非所有多发性硬化患者都应接受MOG-IgG筛查。在诊断有疑问的情况下,结果必须在参考实验室(目前在法国的里昂和克里姆林宫比塞特有实验室)得到确认。儿童的病程通常是单相的,但有可能复发。在成人中,复发的频率似乎高于儿童,估计在5年后超过40%。可能会出现视觉、膀胱/括约肌、认知和(在较小程度上)运动后遗症,但频率远低于NMOSD。
尽管MOGAD是一种罕见的疾病,但预计会造成巨大的经济负担;它有一个严重衰弱的表现,复发后残留残疾。患者可能出现一种或多种视神经炎、脊髓炎、急性播散性脑脊髓炎(ADEM)、大脑单灶性或多灶性缺陷、脑干或小脑综合征或大脑皮质脑炎。症状性发作可归因于视神经、脊髓或大脑的炎症。症状包括失明、眼睛疼痛、头痛、意识模糊、肌肉僵硬或无力、肠道、膀胱或性功能改变以及癫痫发作。与MS不同,MOGAD的神经恶化通常不会在没有复发的情况下进展,这表明有效治疗有可能减少残疾的累积。 根据临床表现的性质、受影响的神经区域以及发作的严重程度、次数和频率,经济负担可能有所不同。误诊和延误治疗也是一个原因,因为MOGAD的异质性表现没有得到很好的识别。
在儿童和成人中,发作用高剂量的IV皮质类固醇治疗,通常非常有效,然后口服减量。在某些情况下,在与中枢神经系统炎性疾病的咨询中心或专家中心讨论后,可能会建议长期免疫活性治疗,特别是在复发时。建议参考/专家中心每年至少进行一次长期随访。在这些预约之间,每6个月对推荐的儿科神经科医师、儿科医师、治疗医师或推荐的神经科医师进行随访。重要的是检查新的发作和并发症的发生,以及在长期治疗的情况下,对治疗的坚持和耐受性。多学科管理是必不可少的,涉及各种医疗保健专业人员。
MOGAD的癫痫发作率为5%-21%,而AQP4抗体阳性的视神经脊髓炎频谱障碍(NMOSD)的发病率为0.4%-1%。MOGAD的3种癫痫表现形式多种多样,虽然多数为局灶性发作,但均为单相发作,MRI表现为孤立性或播散性脑炎。MOGAD并发ON和癫痫的病例已有报道,但均为单相或双相病程,MRI表现为炎性病变。有些患者最初表现正常,但不久后出现更广泛的临床和影像学疾病。
血清蛋白的早期识别需要对MOG抗体进行调查,并及时开始皮质类固醇治疗。开始使用皮质类固醇的可能临界间隔是在症状出现后的48小时内。随后应缓慢口服皮质类固醇,以测试皮质类固醇的依赖性。影响中枢神经系统不同部位的阵发性发作的同步性是相互依存的,而且这两种疾病都对免疫抑制有反应。
参考文献
Banwell B, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023 Mar;22(3):268-282. doi: 10.1016/S1474-4422(22)00431-8.
Giorgi L,et al. French guidelines for the diagnostic and management of MOG antibody-associated disease. Rev Neurol (Paris). 2025 Jun 9:S0035-3787(25)00536-3. doi: 10.1016/j.neurol.2025.04.012.
Aboseif A, et al. Meningitis as an Attack Phenotype of Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. JAMA Neurol. 2025 Jun 16. doi: 10.1001/jamaneurol.2025.1774.
Kleerekooper I, et al. Expanding the phenotype of MOG antibody-associated disease (MOGAD): half a century of epilepsy and relapsing optic neuritis. J Neurol Neurosurg Psychiatry. 2021 Mar;92(3):340-342. doi: 10.1136/jnnp-2020-324323.
Lee L, et al. A narrative review of the economic burden of myelin oligodendrocyte glycoprotein antibody-associated disease and analogous conditions. Front Neurol. 2025 May 30;16:1506465. doi: 10.3389/fneur.2025.1506465.
Liyanage G, et al. The MOG antibody non-P42 epitope is predictive of a relapsing course in MOG antibody-associated disease. J Neurol Neurosurg Psychiatry. 2024 May 14;95(6):544-553. doi: 10.1136/jnnp-2023-332851.

 分享
分享
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言














#临床表现# #MOGAD#
17 举报