
31岁的他反复腰痛、浑身没力气,以为是劳累过度,没想到竟是罕见病作祟
2025-01-13 梅斯罕见新前沿 MedSci原创 发表于陕西省

31 岁李先生因腰痛、皮疹、尿检异常就医,经基因检测等诊断为 Alport 综合征,介绍其临床表现、不同遗传方式及异质性,阐述现有治疗手段,包括经典、新型及终末期肾病治疗等。
31岁的李先生最近总觉得腰部隐隐作痛,并且浑身乏力。他以为只是工作压力大、劳累所致,于是没太当回事。然而,1个月前,他双下肢出现了散在的红色皮疹,针尖大小、高出皮肤,压之不褪色。皮疹虽不痒,但让他有些担忧。
两天前,他在当地医院检查时发现尿检异常:尿蛋白1+、潜血3+,血常规提示白细胞升高。医生初步诊断为 “IgA血管炎”,并建议他进一步检查。随后,李先生来到专科医院,收住入院。
李先生到底怎么了?
李先生,31岁,因反复腰痛、乏力、尿液异常就诊。起初的检查显示尿蛋白和潜血均为阳性,尿红细胞显著增多,同时伴有24小时尿蛋白定量升高。进一步的肾活检显示肾小球基底膜存在节段性厚薄不均和撕裂分层的改变,提示可能存在遗传性肾病。
追问家族史发现,李先生的母亲有尿潜血阳性史,舅舅因肾炎发展为慢性肾衰竭并接受肾脏替代治疗。结合这一线索,医生建议李先生进行基因检测。
基因检测结果揭示了真相。李先生的COL4A5基因存在一处深度内含子变异:chrX:107913428 Intron41[c.3792+1691G>A],为纯合突变,其母亲携带相应的杂合变异。虽然该变异临床意义未明,但结合病理结果,最终诊断为Alport综合征(AS)。
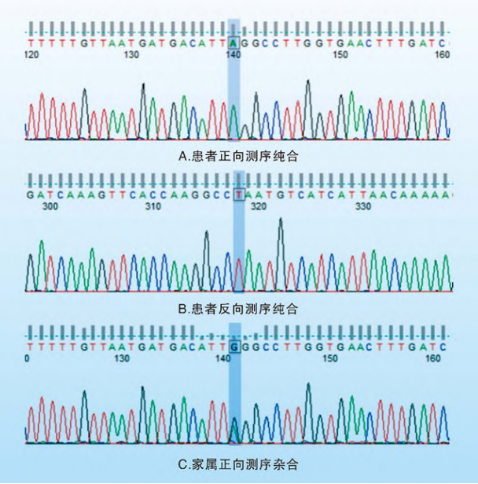
Alport综合征是导致肾衰竭主要的遗传性肾脏病之一
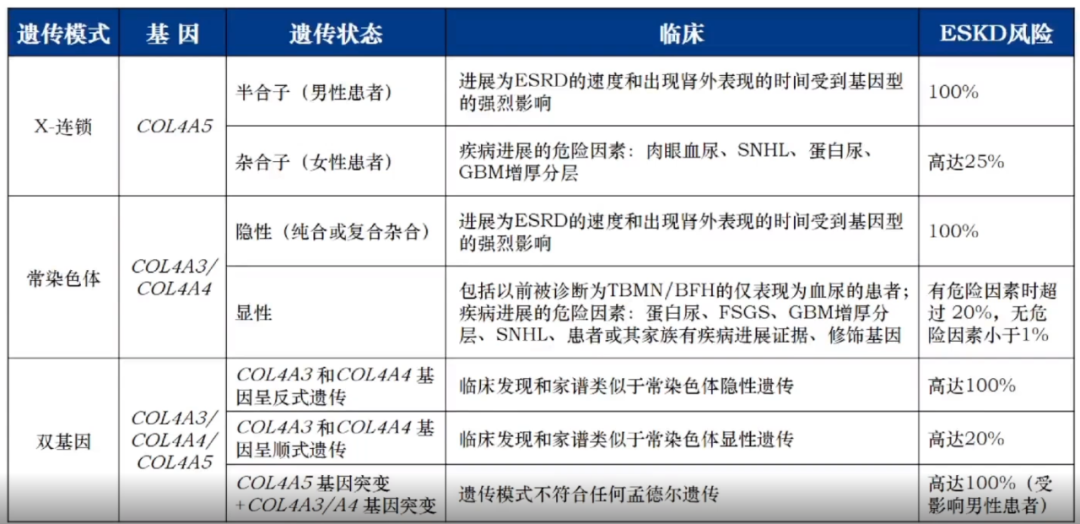
Alport综合征是一种临床表现以血尿、蛋白尿、进行性肾功能减退为特征,部分患者合并感音神经性耳聋(SNHL)、眼部病变等肾外表现的综合征 。该病是由编码Ⅳ型胶原α3α4α5链的COL4A3、COL4A4和COL4A5基因突变导致,
根据不同遗传方式分为X连锁Alport综合征(XLAS)、常染色体隐性Alport综合征(ARAS)、常染色体显性Alport综合征(ADAS)和双基因Alport综合征。不同遗传方式的Alport综合征存在基因型和表型的异质性,但绝大多数Alport综合征患者会进展至终末期肾病(ESKD),是导致肾衰竭主要的遗传性肾脏病之一
临床表现
AS患者的临床表现具有显著异质性,从孤立性血尿到进行性肾功能减退,可能伴随或不伴随肾外表现。以下是AS的主要临床表现:
1. 血尿
血尿是AS最常见的临床表现,多为肾小球源性。XLAS男性和ARAS患者几乎100%存在镜下血尿,约62%的XLAS男性患者和66%的ARAS患者可出现发作性肉眼血尿。
2. 蛋白尿
几乎所有XLAS男性和ARAS患者会随年龄增长发展为持续性蛋白尿,甚至达到肾病范围蛋白尿。XLAS女性和ADAS患者中蛋白尿也较常见,但程度相对较轻。
3. 肾功能减退
XLAS男性患者的肾脏预后较差,90%会在40岁前进展至终末期肾病(ESKD),女性患者中约16%在65岁前进展至ESKD。ARAS患者中位进展至ESKD的年龄为22.5岁,ADAS患者中约39%的蛋白尿患者最终需要肾脏替代治疗,中位进展至ESKD的年龄为67岁。
4. 听力障碍
听力障碍表现为感音神经性听力损失(SNHL),早期累及高频区,需纯音测听检测。听力损失随年龄增长逐渐累及全音域,影响日常交流。XLAS男性患者发生SNHL较女性多且更早。
5. 眼部病变
具有诊断意义的眼部病变包括前圆锥形晶状体和黄斑周围斑点状视网膜病变。前圆锥形晶状体可导致近视度数加深,甚至引起白内障或囊膜穿孔;黄斑病变随肾功能退化而加重,但通常不影响视力。
6. 弥漫性平滑肌瘤病
此表现较少见,常累及胃食管、气管及女性生殖道,可引起吞咽困难、呼吸困难等症状,多与COL4A5基因和COL4A6基因大片段缺失相关。
7. 血管及心脏异常
部分患者伴有青春期颅内动脉瘤、主动脉异常(扩张、夹层或动脉瘤)、二尖瓣脱垂及室间隔畸形等,可能与基底膜结构不稳定有关。
8. 其他罕见表现
AMME综合征是一种少见的综合征,包含Alport综合征、智力低下、中面部发育不良及椭圆形红细胞增多症,由COL4A5基因及相关基因的大片段缺失引起,目前报道较少。
治疗
目前,AS尚无明确的治愈方法,治疗目标主要是延缓肾脏功能的恶化和控制尿蛋白水平。在疾病进展至肾衰竭时,长期透析和肾移植成为延长患者生命的重要手段。
经典治疗
血管紧张素转换酶抑制剂(ACEI)和血管紧张素受体阻滞剂(ARB)是AS治疗的基础药物,能够有效降低尿蛋白水平,减缓肾功能下降。
新型药物与创新疗法
近年来,许多新型药物和创新疗法在延缓AS病程中表现出潜力。钠/葡萄糖协同转运蛋白2抑制剂(SGLT2抑制剂)显示了显著的肾功能保护作用;氨基糖苷类类似物、A型内皮素拮抗剂以及调脂药物和羟氯喹通过不同的机制减少肾纤维化和蛋白尿。此外,抗miR-21、甲基巴多索隆以及基因替代疗法等新兴疗法正在研究中,特别是基因替代疗法,有望从根本上解决因COL4A5基因突变导致的基底膜损伤。
辅助治疗
环孢素A能够减少尿蛋白,但因其潜在的肾毒性,使用时需谨慎。干细胞移植虽然具有修复肾组织的潜力,但尚缺乏足够的临床数据支持其广泛应用。
终末期肾病(ESRD)的治疗
对于进展为终末期肾病的AS患者,长期透析和肾移植是主要治疗选择。其中,肾移植不仅能显著提高患者的生活质量,还可延长生存期,是终末期治疗的首选方案。
参考资料:
1. 万华磊,李莲花,杨馨.COL4A5基因新突变致Alport综合征1例[J].中国中西医结合肾病杂志,2024,25(10):924-925+945.
2.Alport综合征协作组,国家肾脏疾病临床医学研究中心,北京医学会罕见病分会. Alport综合征诊治专家共识(2023版). 中华医学杂志,2023,103(20):1507-1525.
 分享
分享
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#Alport综合征# #COL4A5基因#
32 举报