论文解读|薛澄/梅长林/毛志国教授团队综述:常染色体显性多囊肾病的免疫微环境机制及治疗前景
2025-07-15 Genes and Diseases Genes and Diseases 发表于上海

该综述系统梳理了ADPKD中的免疫细胞类型、炎症因子、补体系统及其相关信号通路的参与机制,并探讨了靶向免疫通路的治疗新策略,为ADPKD临床治疗带来了新的启发。
常染色体显性多囊肾病(ADPKD)是最常见的单基因遗传性肾病,其发病机制长期被认为以基因突变主导。然而,近年来研究表明,ADPKD的病程发展不仅源于PKD1/2突变,还与肾脏局部复杂的免疫微环境密切相关。
近日,来自海军军医大学附属上海长征医院肾脏病科,解放军肾脏病研究所的薛澄/梅长林/毛志国教授团队在本刊发表题为“Immune microenvironment in autosomal dominant polycystic kidney disease”的综述文章。作者包括薛澄博士、李馨茗博士、周晨辰博士、梅长林教授、毛志国教授。该综述系统梳理了ADPKD中的免疫细胞类型、炎症因子、补体系统及其相关信号通路的参与机制,并探讨了靶向免疫通路的治疗新策略,为ADPKD临床治疗带来了新的启发。
1、巨噬细胞极性改变与囊肿生长密切相关
ADPKD早期即出现肾间质免疫细胞浸润,其中以巨噬细胞为主。文章总结发现,M1型巨噬细胞通过TNF-α、IL-1β等促炎因子激活ADPKD囊肿衬里上皮细胞(CLEC)增殖;而M2型巨噬细胞则在晚期促进纤维化进程。肾小管上皮细胞损伤后释放的DAMPs可激活PRR信号通路,进而诱导巨噬细胞活化,构建慢性炎症微环境,促进囊肿持续扩大。
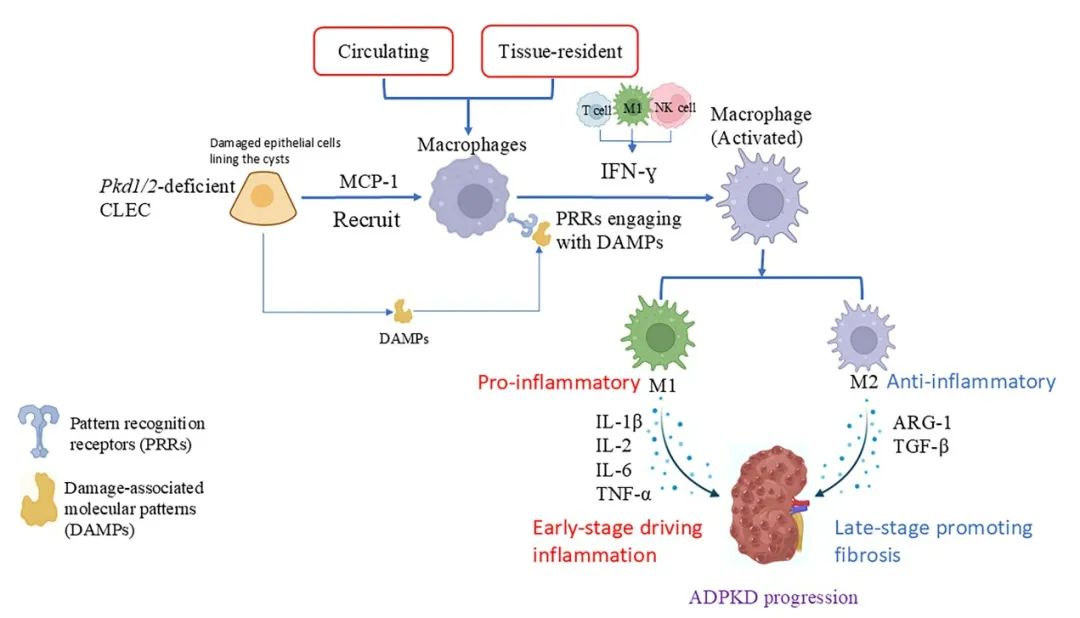
图1 巨噬细胞在ADPKD发病机制中的作用(原文中Figure 1)
肾脏中的巨噬细胞来源于循环中的单核细胞和组织常驻的巨噬细胞。趋化因子(如MCP-1)招募巨噬细胞至受损组织,引导其向两种表型分化:M1型和M2型。巨噬细胞的浸润促进了囊性肾小管上皮细胞(CLEC)的增殖,并推动多囊肾病(PKD)的进展。
2、炎症因子驱动的“囊-免疫”正反馈环
该综述特别强调了ADPKD中炎症因子(如MCP-1、MIF、TNF-α)的关键作用:
MCP-1可招募单核-巨噬细胞,促进其向M2极化,并与GFR下降和肾体积增大密切相关;
MIF促进CLEC增殖和葡萄糖代谢重编程,同时也招募巨噬细胞并通过CD74受体加剧炎症反应;
TNF-α不仅引发免疫细胞浸润,还可通过Akt/mTOR和ERK通路直接刺激囊肿细胞分裂。
这些炎症因子与CLEC之间构成正反馈环,驱动囊肿持续增殖,形成“慢性炎症-代谢失衡-囊肿扩张”的恶性循环。
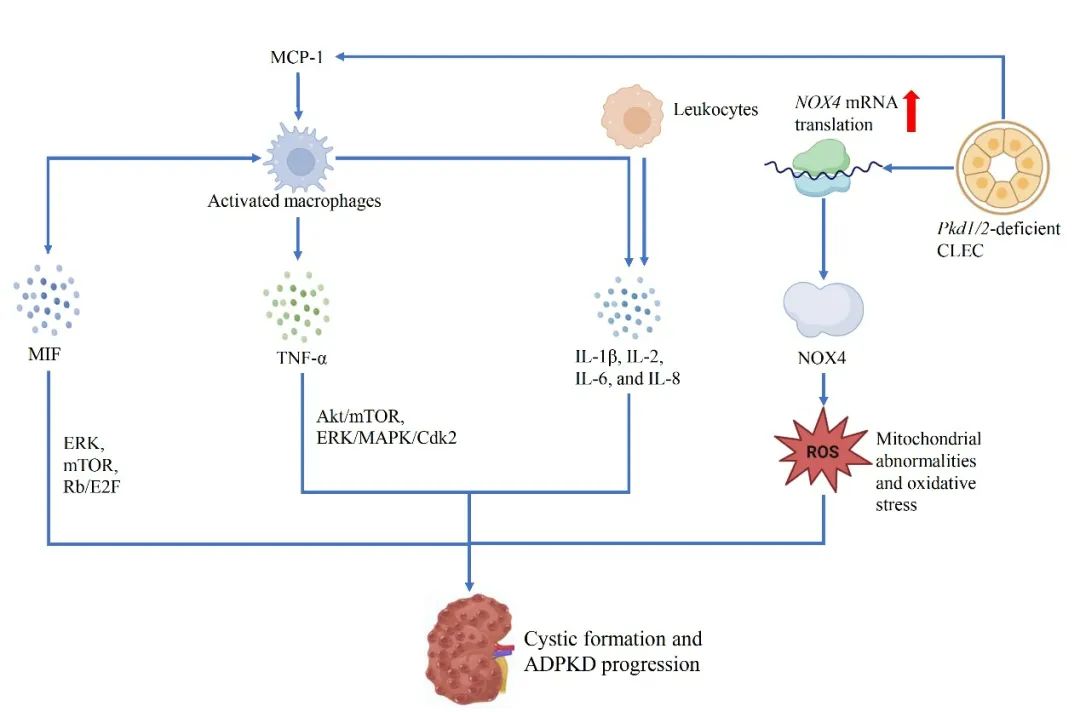
图2 ADPKD中的炎性趋化因子和细胞因子(原文中Figure 3)
CLECs和巨噬细胞分泌的MCP-1可介导巨噬细胞的募集。MIF可通过激活ERK、mTOR和Rb/E2F通路促进巨噬细胞的募集和CLEC的增殖。MIF还可诱导肾小管上皮细胞表达TNF-α,形成一种正反馈回路,助推ADPKD的进展。TNF-α通过Akt/mTOR和ERK/MAPK/Cdk2通路调控CLEC的增殖。多种白细胞介素如IL-1β、IL-2、IL-6和IL-8也在囊肿形成及疾病进展中发挥关键作用。在PKD早期,NOX4表达上调,与线粒体功能异常和氧化应激相关。
3、补体系统异常激活是新兴研究热点
文章指出,补体系统,尤其是替代途径(alternative pathway),在ADPKD中被异常激活。研究者团队在前期研究中发现,C3和CFB在囊肿上皮细胞中表达显著上调。使用替代途径抑制剂迷迭香酸(RMA)可显著抑制囊肿形成,提示补体途径可能是ADPKD治疗的潜在新靶点。
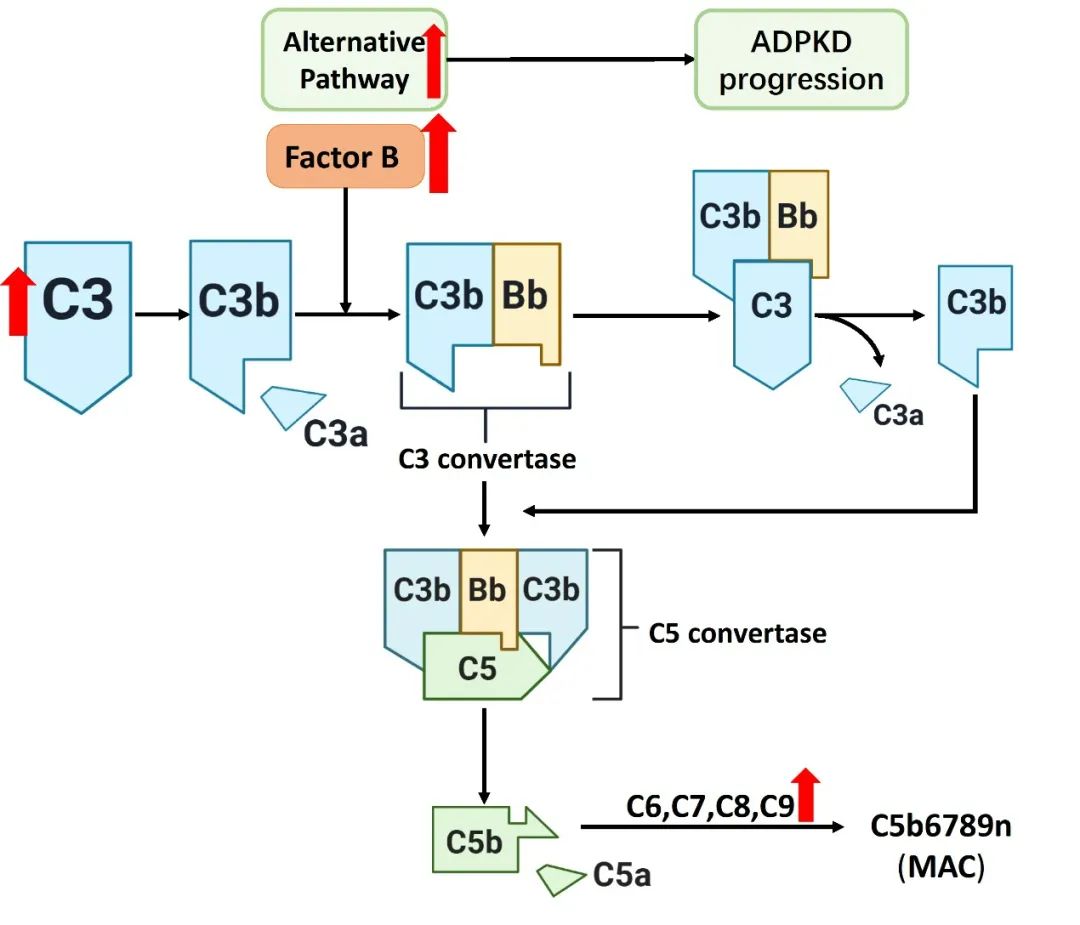
图3 ADPKD中活化的补体系统(原文中Figure 4)
补体系统,尤其是替代途径在ADPKD发病机制中起重要作用。在ADPKD患者中,C3显著激活,因子B(factor B)和C9的表达水平升高。
4、炎症相关信号通路贯穿疾病全过程
NF-κB通路作为促炎核心通路,在ADPKD中被显著激活,并调控多种细胞因子的转录;
JAK-STAT通路在CLEC中参与细胞增殖、免疫激活及巨噬细胞极化;
Nrf2通路因其抗氧化与抗炎作用,在ADPKD后期被抑制,其激活可改善肾功能并减缓病情进展。
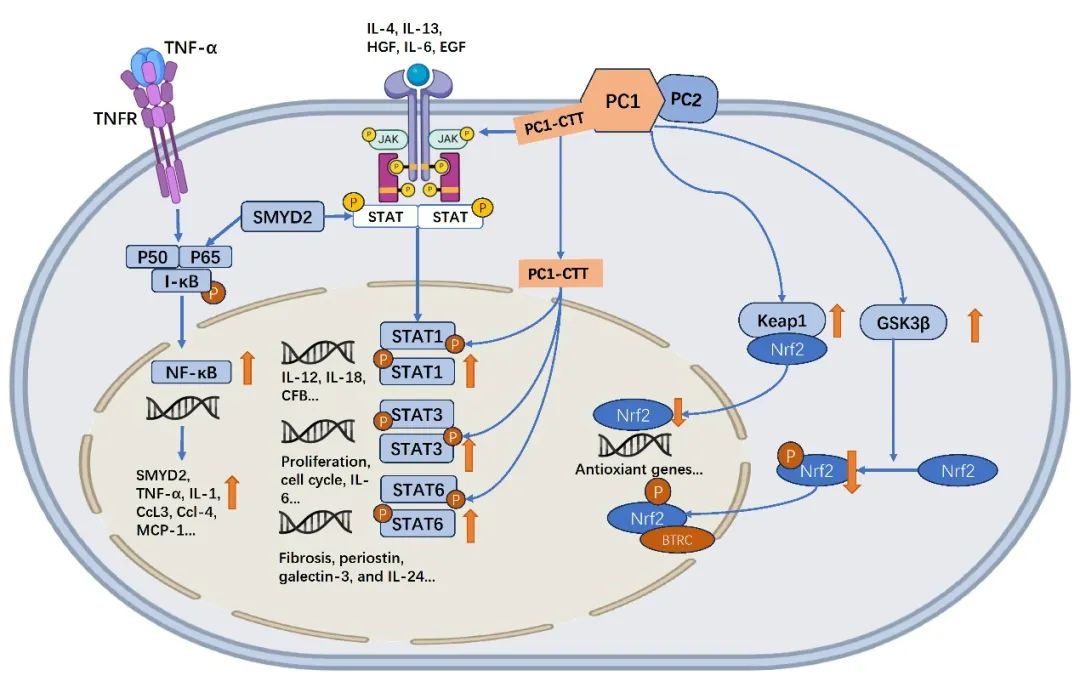
图4 ADPKD中的炎症信号通路(原文中Figure 5)
在ADPKD中,NF-κB显著活化,驱动如TNF-α、IL-1、CCL2/3/4、MCP-1和C3等促炎因子和趋化因子的转录。多囊蛋白1(PC1)可激活PKCα介导的NF-κB信号通路。SMYD2负责激活STAT3和NF-κB的p65亚基。JAK/STAT通路在ADPKD中促进囊肿上皮细胞的增殖及免疫反应。PC1的C端尾(PC1-CTT)增强STAT1、STAT3和STAT6的活性,并通过JAK2/STAT1轴调控补体因子B(CFB)表达。KEAP1是Nrf2的主要负调控因子,通过与Nrf2结合促进其泛素化并由蛋白酶体降解。GSK3β通过磷酸化Nrf2加强其降解,进一步抑制其信号传导。
5、补体系统异常激活是新兴研究热点
该综述全面梳理了当前针对ADPKD免疫微环境的多种潜在治疗策略,包括:
早期干预:雷公藤酯(triptolide)、白藜芦醇(resveratrol),通过抑制NF-κB、STAT3等通路,延缓囊肿形成;
中期干预:使用TNF-α抑制剂(如Etanercept)、S1P通路抑制剂(如FTY720)、补体抑制剂(如RMA);
晚期干预:靶向巨噬细胞耗竭、免疫检查点(如PD-1/CTLA-4)联合抑制等策略,以改善晚期ADPKD炎症环境。
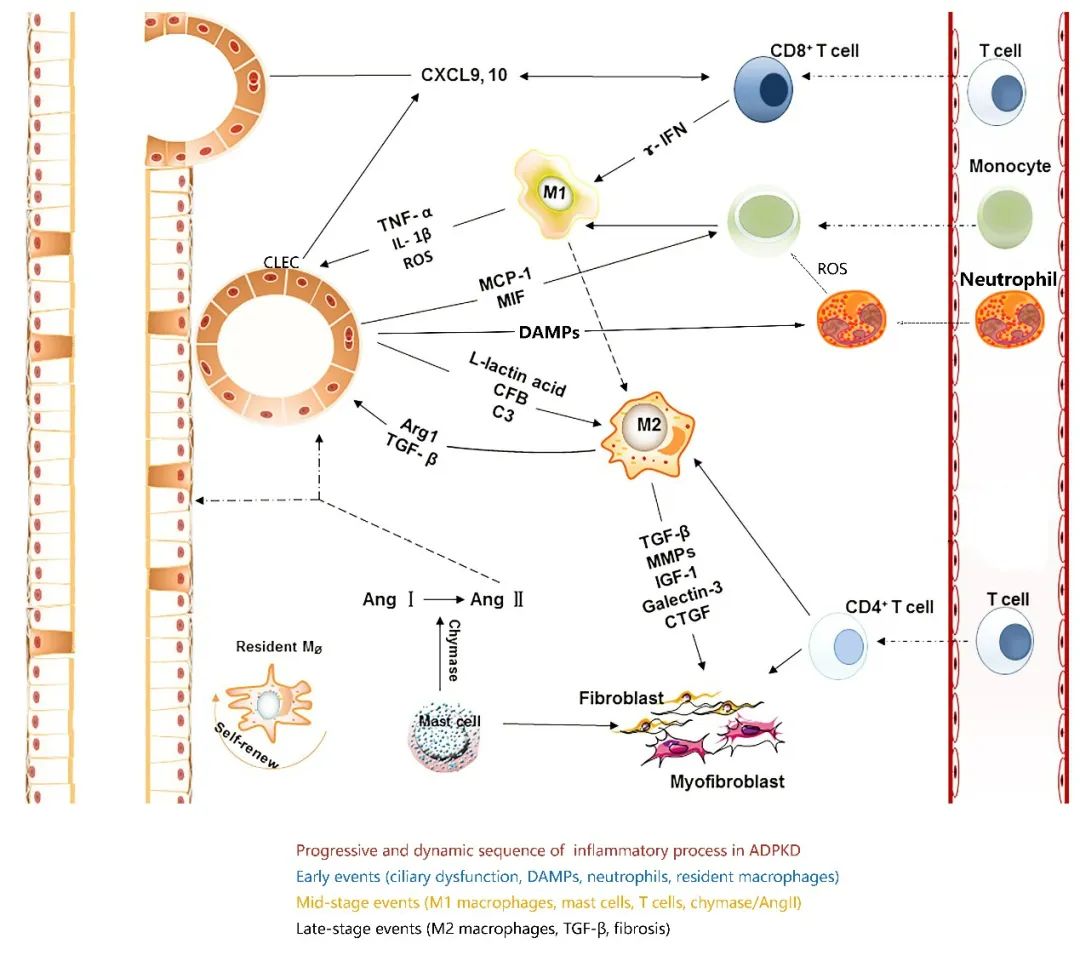
图5 常染色体显性多囊肾病中的免疫细胞(原文中Figure 2)
6、ADPKD中的免疫细胞及治疗策略
活化的CD8⁺ T细胞释放干扰素-γ(IFN-γ),促进巨噬细胞向促炎性表型(M1型)极化。M1型促炎性巨噬细胞分泌细胞因子,如肿瘤坏死因子-α(TNF-α),从而促进囊性上皮细胞(CLECs)的增殖。CLECs会分泌乳酸,进一步刺激巨噬细胞向促纤维化表型(M2型)转化。此外,活化的CD4⁺ T细胞也促进巨噬细胞向M2型极化。M2型巨噬细胞高表达精氨酸酶1(ARG1),可促进囊肿增大。M2型巨噬细胞分泌多种促纤维化细胞因子,如转化生长因子-β(TGF-β)、结缔组织生长因子(CTGF)、血小板源性生长因子(PDGF)和类胰岛素生长因子(IGF),刺激成纤维细胞和肌成纤维细胞的活化,进而促进肾脏纤维化的进展。肥大细胞释放胰凝乳蛋白酶(chymase),可将血管紧张素I(Ang I)转化为血管紧张素II(Ang II),而Ang II可促进肾组织损伤及纤维化的发展。
7、总结
本综述强调,ADPKD不仅仅是结构突变引起的囊肿疾病,更是一个由免疫-炎症-代谢共同作用的系统性疾病。深入理解ADPKD中免疫微环境的构建过程,不仅有助于揭示疾病机制,也为开发精准、阶段性干预手段提供理论基础。未来,靶向炎症、补体和免疫检查点的治疗策略,有望成为ADPKD干预的新突破口。
文章来源
免费全文下载链接:
https://www.sciencedirect.com/science/article/pii/S2352304225001837
引用这篇文章:
Xue C, Li X, Zhou C, et al. Immune microenvironment in autosomal dominant polycystic kidney disease. Genes Dis. In press.

 分享
分享
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#常染色体显性多囊肾病# #免疫微环境#
4 举报